Agentic AI Is Coming to the SaMD Lifecycle
A phase-by-phase technical analysis of agent patterns, guardrail architectures, and human-in-the-loop gates across the IEC 62304 lifecycle.
Designing, building, and shipping Software as a Medical Device (SaMD) must comply with some of the most demanding regulatory frameworks in any industry. From early concept work to FDA 510(k) submissions, from IEC 62304 lifecycle management to post-market CAPA processes, every step of the SaMD pipeline (Figure 1) demands extraordinary rigor in documentation, traceability, and quality assurance. It’s grueling work. And most of the time, it’s not the clinical or engineering decisions that slow you down. It’s the documentation.
That’s why I’ve been paying close attention to Agentic AI, a class of autonomous systems that don’t just answer questions but actually plan, execute, and verify multi-step tasks across complex workflows. This is not theoretical anymore. Deloitte reports that over 80% of healthcare executives expect agentic AI to deliver significant value across clinical and operational functions in 2026, and 61% are already building or budgeting for it [1].
The question for those in regulated medical software is no longer whether this technology will reach our world. It’s how we harness it responsibly at every phase of the SaMD development lifecycle.

Figure 1: The SaMD Development Lifecycle with Agentic AI Augmentation
TL;DR
Agentic AI can augment all six phases of the SaMD lifecycle, from ideation and user needs through post-market surveillance, by automating documentation, traceability, and process coordination tasks that consume a disproportionate share of engineering effort today.
The agent capabilities are not the hard part. The guardrail architecture, which includes hallucination detection, completeness validators, pre-execution plan review, and human-in-the-loop gates, is the core engineering challenge in regulated contexts.
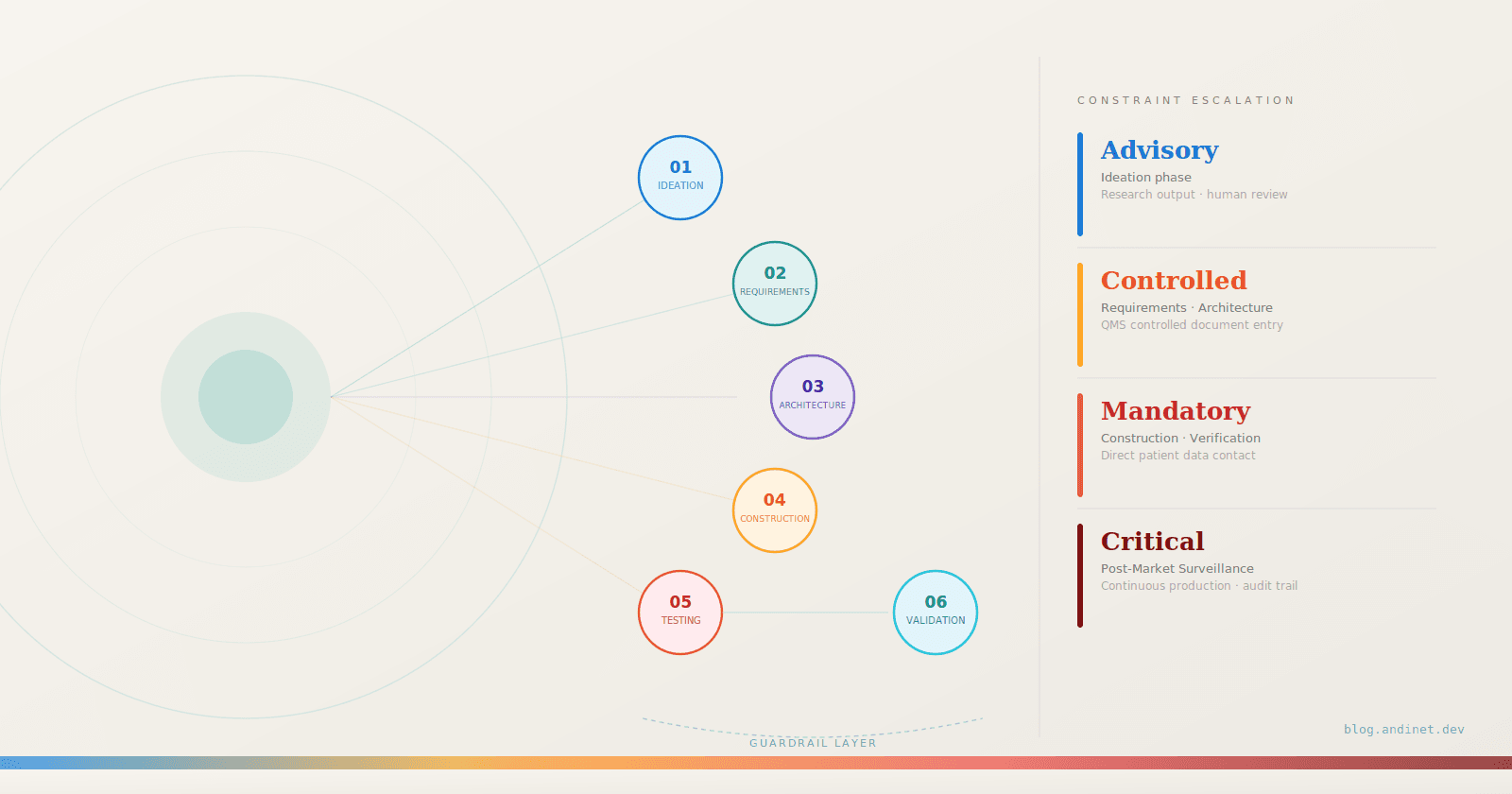
Guardrail constraint levels escalate from Advisory at ideation to Critical at post-market, directly tracking the proximity of agent outputs to patient-facing clinical decisions and regulatory artifacts.
Most agent patterns described here are conceptual designs that illustrate what is achievable, not systems running in production. The distinction matters, and the final section addresses it directly.
What Makes Agentic AI Different?
Most of us in MedTech are already familiar with AI and machine learning. We’ve seen it embedded in diagnostic imaging tools, clinical decision support, and predictive analytics. The FDA has now authorized over 1,450 AI-enabled medical devices as of mid-2025 [2]. But agentic AI is a different animal entirely.
Traditional AI responds to a single prompt with a single output. Agentic AI systems perceive, plan, act, and verify autonomously across multi-step workflows. They maintain context, use tools, call APIs, coordinate with other agents, and loop back to correct their own work. Think of it less as a chatbot and more as a capable junior team member who can execute a defined process end to end, flagging exceptions for human review along the way.
For those of us who live within the SaMD lifecycle, a pipeline defined by IEC 62304 and governed by rigorous Quality Management Systems matters a lot. Here’s how agentic AI can augment each phase of the process our team has followed for years.
Agentic AI Terminology
Before walking through each phase, a quick primer on terminology. In agentic AI, an agent is a decision-making layer that takes a goal, develops a plan, invokes tools, and adapts based on results (as shown in Figure 2). Skills are reusable, domain-specific capabilities the agent can invoke (think of them as functions it has been taught). Tools are external software APIs, databases, or services that the agent can call during execution to provide additional capabilities. Commands are discrete instructions that the agent issues to tools or sub-agents. And orchestration is how you coordinate multiple agents, route tasks, and enforce policies across the workflow. In regulated medical software, guardrails are the constraints, validators, and human-in-the-loop checkpoints that prevent the agent from producing or executing anything that violates your QMS, your risk framework, or patient safety requirements. In this domain, the guardrails aren’t an afterthought. They’re the product.
A note before we dive in: the six-phase patterns that follow describe how I believe agentic AI could augment each stage of the SaMD lifecycle. Some elements exist in commercial tools today; most are conceptual designs that illustrate the shape of what’s possible, not systems running in production. I return to this caveat in more depth at the end of the article.

Figure 2: Anatomy of an Agentic AI System for Regulated Software
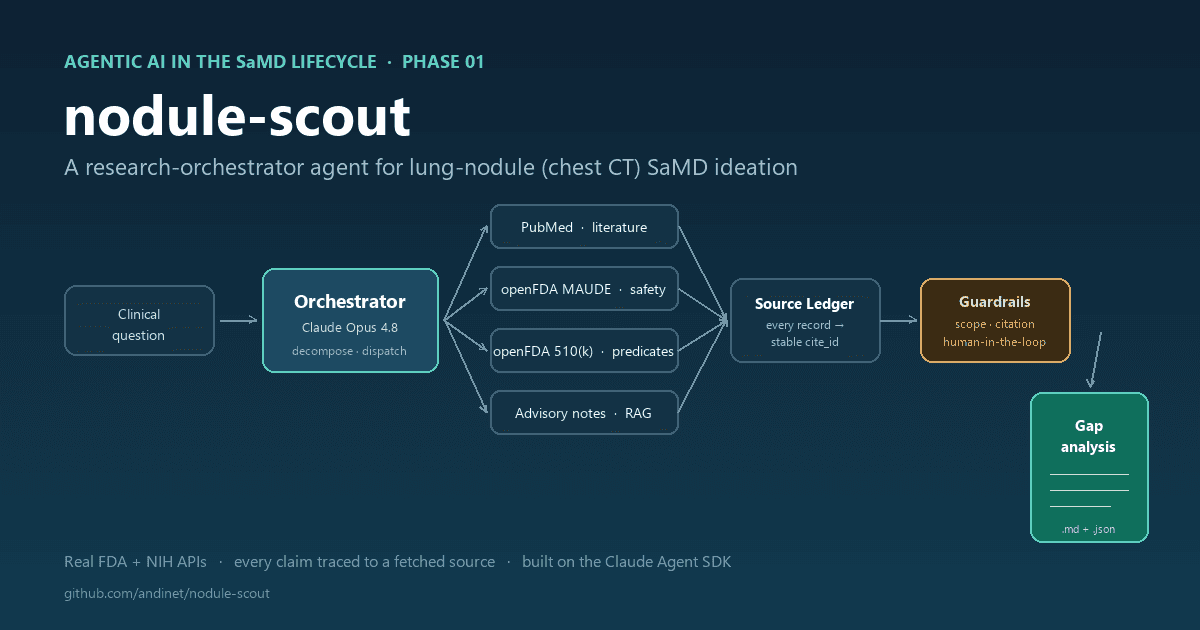
Phase 01: Ideation and User Needs
THE PROPOSED AGENT PATTERN: Imagine a research orchestrator agent that would decompose a broad clinical question (“What are the unmet needs in breast MRI triage software?”) into sub-tasks and dispatch them to specialized tool-calling agents. One sub-agent would have a PubMed search skill that would retrieve recent clinical literature. Another would have an MAUDE query skill that would mine the FDA’s adverse event database for reported software failures in similar SaMD products. A third would have a 510(k) summary skill that would pull predicate device labeling from the FDA database. The orchestrator would collect results, run them through a retrieval-augmented generation (RAG) pipeline grounded in your company’s internal clinical advisory board notes, and produce a structured gap analysis document.
GUARDRAILS AND EVALUATION: This is research output, not a regulatory submission, but it still needs constraints. The key guardrail here is source attribution and hallucination detection. Every claim in the gap analysis must cite a specific PubMed ID, MAUDE report number, or 510(k) summary. A validator agent runs post-generation, checking that every cited source actually exists and that the claim reasonably follows from the source text. It’s worth noting that hallucination detection in RAG systems is itself an active research area with meaningful false-negative rates, so the threshold for flagging outputs for human review should be conservative rather than aggressive. You also need a scope constraint that prevents the agent from drifting into clinical domains outside the intended use you defined, because a gap analysis that wanders into neurology when you’re building a breast imaging tool is worse than useless.
Phase 02: Requirements Gathering and SRS
THE PROPOSED AGENT PATTERN: Imagine a requirements engineering agent with two core skills. First, a need-to-requirement mapping skill that would take each user need statement and generate candidate technical requirements following your SRS template format, complete with requirement IDs, priority levels, and risk classification tags per ISO 14971. Second, a traceability matrix skill that would auto-populate a bidirectional traceability matrix linking each user need to its derived requirements, and forward-link placeholders for design inputs, code modules, and verification tests. The agent would use structured output schemas (enforced JSON or XML) so the output is machine-parseable and can be imported directly into your requirements management tool, whether that’s DOORS, Jama, or even a controlled spreadsheet in your QMS. Products like Ketryx already demonstrate that automated traceability generation integrated with tools like Jira and GitHub is commercially viable.
GUARDRAILS AND EVALUATION: This is where constraints get serious, because a bad requirement can cascade through your entire submission. The first guardrail is a completeness validator that cross-checks every user need against the generated requirements and flags any need that has zero derived requirements. The second is a regulatory consistency checker, essentially a RAG pipeline grounded in IEC 62304 clause text and your company’s SOP templates, that reviews each requirement for proper structure: is it testable? Is the acceptance criterion measurable? Does it use “shall” language correctly? A third possibility, though one that remains more aspirational than production-ready today, is a predicate device comparator that compares your draft SRS against the predicate device’s labeled indications for use and flags any requirement that would constitute a new intended use not covered by your regulatory pathway. This kind of nuanced regulatory judgment is an area where experienced RA professionals still have a significant edge over current AI capabilities. Every output goes through a human-in-the-loop approval gate before it enters your QMS as a controlled document. The agent proposes. The regulatory and clinical team disposes.
Phase 03: Architecture and Software Design
THE PROPOSED AGENT PATTERN: Picture a design architect agent equipped with domain-specific skills for the open-source medical software ecosystem. For example, such an agent would have deep context for radiology software applications (via RAG over the ITK, VTK, and 3D Slicer documentation and source code) on standard architectural patterns in radiological software. It would know that your DICOM ingestion layer will likely use DCMTK or pydicom for parsing, that your image processing pipeline will be built on ITK filters for registration and segmentation, that your 3D visualization will use VTK’s rendering pipeline, and that your application framework might be a 3D Slicer loadable module following the Model-View-Controller pattern with MRML nodes for data management. If you’re deploying a deep learning inference component, it would understand the MONAI ecosystem: MONAI Core for training, MONAI Deploy App SDK for packaging the model as a MAP (MONAI Application Package) with DICOM I/O operators. The agent would use these skills to draft a Software Design Document that describes module decomposition along the lines your codebase actually follows. It could classify your SOUP (Software of Unknown Provenance) components and flag that ITK 5.x, VTK 9.x, and the specific MONAI model bundle each require documented risk assessments under IEC 62304. It could propose a cybersecurity threat model for your DICOM data pipeline, identifying where PHI is handled and where your PACS integration introduces an attack surface.
GUARDRAILS AND EVALUATION: The design phase guardrails operate at two levels. The first is architectural constraint validation: a rule-based validator that checks the proposed design against your SRS and flags any requirement that has no corresponding design element, or any design element that traces to no requirement. This catches scope creep and orphan modules before a single line of code is written. The second is a SOUP risk evaluator that queries known vulnerability databases (CVE, NVD) for each third-party library the design depends on and flags any component with unresolved critical vulnerabilities. This matters enormously for cybersecurity compliance, as the FDA can now refuse to accept submissions that don’t include a Software Bill of Materials (SBOM) with vulnerability analysis. A third guardrail is a design-review preparation tool that generates probing questions such as “What happens if the DICOM series has inconsistent slice spacing?” or “How does the system behave when the MONAI inference returns a confidence score below the clinical threshold?” I want to be clear that these supplements do not replace a formal design review. Real design reviews involve domain-expert judgment, failure mode analysis, and regulatory context that current AI systems handle inconsistently. But the preparation work alone can save significant time. Every design output requires sign-off from both the software lead and the quality team before it becomes a controlled document.

Figure 3: Construction Phase: Agent, Skills, and Guardrail Stack for AI-enabled Medical Imaging Software
Phase 04: Construction and Coding
THE PROPOSED AGENT PATTERN: This is where I see the most immediate impact, because the open-source medical imaging toolkits we rely on are massive, well-documented codebases that are well-suited for agentic code generation. The pattern here would be a coding agent with specialized skills loaded via context: a 3D Slicer module scaffolding skill, an ITK pipeline construction skill, a VTK visualization skill, and a MONAI training/deployment skill. Each skill is essentially a RAG-grounded prompt template backed by the relevant codebase documentation and thousands of existing module examples.
Imagine giving a coding agent a command like “Implement SRS requirement REQ-IMG-042: the system shall segment liver parenchyma from contrast-enhanced CT with Dice coefficient >= 0.92.” Its orchestration layer would break this into sub-tasks. It would scaffold a new 3D Slicer loadable module that inherits from qSlicerLoadableModule, set up the MRML node observers, and implement the module Logic class. For the segmentation itself, it would wire in a MONAI-trained UNet model using the MONAI Bundle format, generate the inference pipeline with the appropriate transforms (LoadImage, Spacing, Orientation, NormalizeIntensity), and use MONAI’s sliding window inference for large 3D volumes. It would generate the VTK rendering code to display the segmentation as a 3D surface overlaid on the original DICOM volume. It would know the correct CMake configuration to link against the Slicer superbuild. And it would produce a Python test harness that maps directly to REQ-IMG-042.
For the deep learning training pipeline, the agent could generate a properly structured MONAI workflow using CacheDataset for efficient data loading, domain-appropriate augmentation transforms (RandGaussianNoise, RandAffine, Rand3DElastic), DiceFocalLoss to handle class imbalance, and the Novograd optimizer. It could also generate the MONAI Deploy application packaging code that wraps the inference model as a MAP with DICOM input/output operators, making it ready for clinical PACS integration.
GUARDRAILS AND EVALUATION: Construction is where guardrails need to be the tightest, because code is the artifact that directly touches patient data and clinical decisions. The first guardrail is pre-execution plan review. Before the coding agent writes anything, it outputs a structured plan describing which files it will create, which libraries it will import, and which requirements it will address. A guardian agent evaluates this plan against the Software Design Document and blocks execution if the plan introduces dependencies not in the approved SOUP list, touches modules outside the agent’s assigned scope, or proposes an architecture that deviates from the SDD. This is the “intervene at planning, not at execution” pattern that recent guardrail research emphasizes for high-stakes domains.
The second guardrail is output validation. Every code artifact the agent produces goes through a multi-layer check: static analysis for coding standard compliance, automated build verification (does it compile against the Slicer superbuild?), unit test execution with coverage thresholds, and a traceability check confirming every function maps to a requirement. The third guardrail is the most critical for regulated software: no auto-merge. The coding agent generates a pull request with full documentation, test results, and traceability links. A human developer reviews, approves, and merges. The agent accelerates the work. It never has the final say.
Phase 05: Testing and Verification
THE PROPOSED AGENT PATTERN: Imagine a verification orchestrator agent that would read the traceability matrix and auto-generate a comprehensive test plan. It would dispatch sub-agents with specialized testing skills. A DICOM edge case agent would create synthetic test fixtures covering varied slice thicknesses, different transfer syntaxes, vendor-specific private tags (if you’ve ever dealt with Siemens vs. GE DICOM headers for diffusion MRI, you know how varied those can get), and missing optional fields. A model evaluation agent would generate evaluation scripts that compute Dice scores, sensitivity, specificity, and AUC against annotated ground truth datasets, then compare results against the acceptance criteria defined in the SRS. A regression test agent would build and run suites that verify the ITK segmentation pipeline produces consistent output across image modalities, resolutions, and patient populations. A performance test agent would benchmark VTK rendering latency on target hardware configurations. The orchestrator would collect all results and generate formatted verification reports in the template your QMS requires.
GUARDRAILS AND EVALUATION: Verification guardrails are about ensuring the tests themselves are trustworthy. The first constraint is test completeness validation: a validator that compares the test plan against the traceability matrix and flags any requirement with zero associated test cases. This is a common audit finding, and catching it automatically before the verification phase closes is worth its weight in gold. The second is test independence verification: the agent must not generate tests that are trivially tautological (testing that a function returns the value it was hardcoded to return). A reviewer agent evaluates each test for meaningful assertion logic. The third guardrail is a golden dataset integrity check: before running model evaluation, the agent verifies that the ground truth dataset hasn’t been modified since it was locked in the validation plan, by checking cryptographic hashes. For ML-based SaMD components, the FDA expects documented evidence of dataset integrity. The final gate: all verification reports go through a formal review by the quality team. The agent produces the evidence. The human signs the DHF (Design History File).
Phase 06: Validation and Post-Market Surveillance
THE PROPOSED AGENT PATTERN: Validation and post-market are where agentic AI shifts from batch execution to continuous autonomous monitoring. For the validation phase, a clinical evaluation agent drafts the initial validation plan by pulling from FDA guidance on SaMD clinical evaluation (the IMDRF framework), identifying appropriate performance metrics for your device classification, and recommending study designs based on your risk category. But the real power is in post-market. Here I envision deploying a surveillance multi-agent system: one agent monitors deployed model inference metrics in production (throughput, confidence distributions, error rates); a second agent runs statistical drift detection comparing incoming data distributions against the training set baseline; a third agent watches the MAUDE database and relevant literature for reports involving your device class. These agents communicate through an event bus. When the drift detection agent flags a distribution shift that exceeds your Predetermined Change Control Plan (PCCP) thresholds, it triggers the documentation agent to generate a change assessment record and the notification agent to alert your quality team.
I want to be transparent that this multi-agent surveillance architecture is a proposed design, not a description of something running in production today that I know of. No published implementation of this specific pattern exists in regulated medical software. But the individual components (drift detection, MAUDE monitoring, automated documentation) are all technically feasible, and the value proposition for continuous post-market compliance is compelling enough that I expect early implementations within the next two to three years.
GUARDRAILS AND EVALUATION: Post-market guardrails are arguably the most consequential because they would run continuously, autonomously, in production, touching real patient outcomes. The paramount constraint is the agent can observe and report, but it cannot modify the deployed model or its clinical behavior. That boundary is non-negotiable. The monitoring agents are read-only with respect to the production system. Any model update, retraining decision, or field safety corrective action requires human authorization through your CAPA process. The second guardrail is alert fatigue management: threshold tuning so the drift detection agent doesn’t flood your quality team with noise. This requires calibrated sensitivity/specificity for the detection algorithm itself, which means the guardrail needs its own evaluation framework. The third is audit trail completeness: every agent action, every tool call, every decision point is logged immutably. In a regulatory audit, you need to reconstruct exactly what the system observed, when it flagged an issue, what was reported, and who acted on it. The NIST AI Risk Management Framework’s emphasis on traceability for every decision and tool call maps directly to this requirement. If you can’t show the audit trail, the autonomous monitoring is a liability, not an asset.

Figure 4: Guardrail Severity Across the SaMD Lifecycle
Here’s the pattern that runs through every phase: the agentic system has agents, skills, and tools to do the heavy lifting. But at every step, there are guardrails that constrain what the agent can do, validators that evaluate what it produced, and human-in-the-loop gates that control what enters your QMS as a controlled artifact (Figure 4). In regulated medical software, the guardrail architecture is not a safety net. It is the core engineering challenge. Get it right, and you have a system that produces auditable, traceable, compliant outputs at machine speed. Get it wrong, and you’ve automated your way into a regulatory nightmare.
A Word of Caution: What This Article Does Not Claim
The agent patterns described across these six phases represent a technically coherent design space, but they sit at different points on the spectrum between commercially available today and not yet demonstrated in production. Coding agents that scaffold module templates, documentation generators that populate SRS fields, and drift detection pipelines are all capabilities that exist in some form in current tooling. The full multi-agent surveillance architecture described in Phase 06, with autonomous orchestration across monitoring, drift detection, MAUDE surveillance, and documentation generation, is a proposed design. I am not aware of a published implementation of that specific pattern in a regulated medical software environment as of this writing.
There is also a meta-problem that most discussions of agentic AI in MedTech either ignore or treat as a footnote: if you deploy AI agents as part of your SaMD development process, those agents may themselves require validation as software tools under your QMS. The FDA expects that development tools affecting product quality are validated. An agentic system that generates SRS requirements, writes code, or produces verification reports is exactly that kind of tool. The guardrail architecture described in this article addresses the outputs produced by those agents. It does not address the harder question of how you validate the agent platform itself as a controlled development environment — with documented intended use, qualification testing, and change management procedures. That question does not yet have a definitive answer in the industry, and anyone deploying these systems in a GxP context should think about it carefully.
Finally, the argument that agentic AI addresses the documentation and traceability burden in SaMD development is grounded in direct experience, but the specific ratio of documentation effort to clinical and engineering effort varies significantly across organizations, device classifications, and QMS maturity. The opportunity is real. The implementation complexity is also real. Both things are true simultaneously.
The Pattern That Matters
The six-phase patterns in this article share a common structure: agents and skills to execute the work, tools to extend their reach, and a guardrail stack that escalates in constraint level as outputs move closer to patient-facing decisions and controlled regulatory artifacts. As the severity table in Figure 4 shows, the progression runs from Advisory at ideation, where outputs are research documents subject to human review, through Controlled at requirements and architecture, Mandatory at construction and verification, where code directly touches patient data, and Critical at post-market, where autonomous monitoring runs continuously against a production clinical system.
That escalation is not arbitrary. It reflects a straightforward principle: the closer an agent output is to a controlled QMS artifact or a patient outcome, the less latitude the agent has and the more explicit the human authorization requirement becomes. Downstream quality and regulatory teams will recognize this pattern immediately as it maps directly to the risk-based thinking embedded in IEC 62304, ISO 14971, and FDA's own guidance on software change control. The guardrail architecture described here is not a novel compliance framework. It is an application of existing regulatory logic to a new class of development tooling.
The SaMD lifecycle, as currently practiced, allocates a significant share of skilled engineering time to documentation, traceability, and process coordination. Agentic AI does not change what the work requires. It changes who — or what — does the mechanical portions of it, under what constraints, and with what level of human oversight at each transition. The companies that instrument this correctly will move faster through their development cycles without accumulating the compliance debt that typically results from moving fast in regulated software. That is the practical case for the investment. The guardrail architecture is what makes it defensible.
References
[1] Deloitte. "Many health care leaders are leaning into agentic AI as adoption hurdles ease." 2026 US Health Care Outlook Survey, February 2026. Link
[2] The Imaging Wire. "Numbers from the FDA Show Radiology Is Maintaining Its Lead." March 11, 2026. Link
[3] Papademetris, X. "Introduction to Medical Software." Yale University, Coursera. Link
Disclaimer
The views, opinions, and technical perspectives expressed in this article are entirely my own and do not represent, reflect, or imply the positions, strategies, or endorsements of my employer or any organization I am affiliated with.